Марат Хамадеев
Марат Хамадеев
Самообучение графов на основе дескрипторов помогло предсказать токсичность молекул
Людям известно более сотни химических элементов, уложенных в периодическую таблицу. Они могут объединяться в молекулы, чьё разнообразие уже не поддается счёту. Каждая молекула характеризуется элементным составом и структурой, которые по сложным квантовомеханическим правилам определяют её биохимические свойства. Понимание этих свойств важно для множества областей человеческой деятельности, в первую очередь, для поиска новых лекарств.
Традиционным подходом к выявлению этих свойств являются лабораторные эксперименты с растворами, из-за чего он называется «мокрым» (от английского «wet lab»). Последние полвека в противовес «мокрой» активно развивается «сухая» молекулярная биология, в рамках которой ученые предсказывают свойства химический соединений с помощью компьютеров.
За это время методы вычислительной биологии сильно эволюционировали, и сегодня исследователи связывают свои надежды с глубоким обучением. В задаче о предсказании свойств молекул хорошо себя показали графовые нейронные сети, поскольку молекулы вполне естественным образом можно представить в виде некоторого графа: в его узлах располагаются атомы, а рёбра соответствуют химическим связям.
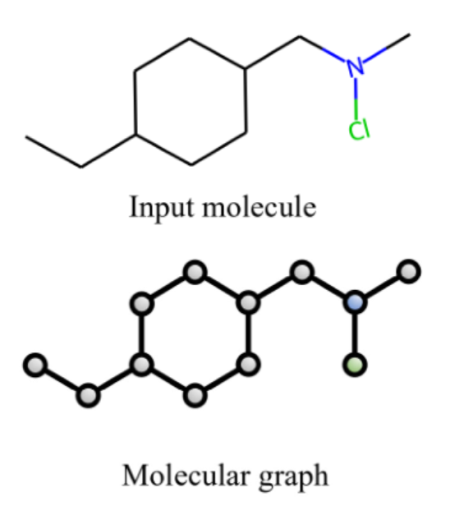
Представление молекулы с помощью графа
Чтобы начать глубокое обучение обычно требуется большой объем размеченных данных: информация о структуре молекулы, входящих в нее атомов и связей и конкретное значение свойств этой молекулы, например, токсичности, которые получают в ходе «мокрых» экспериментов. Тем не менее, неразмеченных данных гораздо больше, чем размеченных и собирать дополнительную разметку может быть очень дорого. Поэтому сейчас популярным становится самообучение (Self-supervised Learning, SSL), в котором в качестве разметки используются внутренние свойства данных.
Эта техника стартует с предобучения на неразмеченных данных, задачей которого является самостоятельное выучивание представления объектов, после чего модель можно использовать в режиме обучения с учителем на новом малом объеме размеченных данных. Известные на сегодня подходы к самообучению графовых нейронных сетей применительно к молекулам, либо используют маскирование и предсказание атомных свойств при предобучении на уровне узлов, либо собирают информации в подграфах на уровне молекулярных подструктур.
Группа ученых из ВШЭ, AIRI и МИСиС под руководством Ильи Макарова уверена, что эти подходы не оптимальны, поскольку вспомогательные задачи на уровне узлов не сохраняют полезные знания предметной области, а стыковка методов на основе мотивов с задачами на уровне узлов требует больших вычислительных ресурсов. Вместо этого команда разработала подход к самообучению графов на основе молекулярных дескрипторов.
Молекулярными дескрипторами называют некоторые числовые значения, которые выражают химические свойства через элементный состав и структуру. Молекулу можно разбить на подструктуры, содержащие один или несколько атомов, согласно их биологической значимости — в данном случае по наличию электронов, формирующих π-связь. Для описания таких центров существует специальный дескрипторный язык, известный как фрагментарный код суперпозиции подструктур (fragmentary code of substructure superposition, FCSS). Дескрипторные центры могут состоять из гетероатомов, атомов углерода с двойной или тройной связью и ароматических фрагментов. Поскольку ароматичность может определяться свойствами атомов или связей (если атом или связь являются ароматическими), авторы сосредоточились только на первых двух случаях и выделили 17 типов дескрипторных центров из всего FCSS.
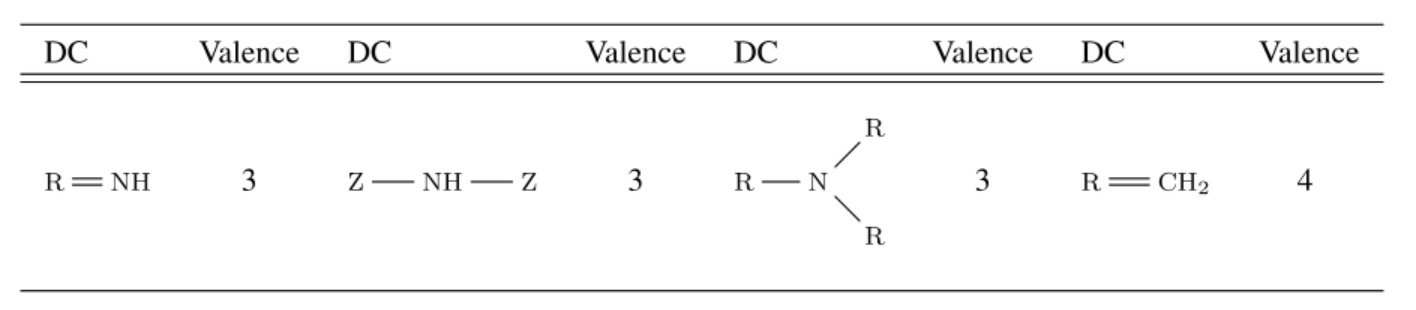
4 из 17 дескрипторных центров, которые выделили ученые. Z обозначает любой атом, R — любой атом кроме водорода
С помощью таких дескрипторных центров исследователи усовершенствовали задачу предсказания. Обычно она решается с опорой на кодирующий вектор, который представляет 119 химических элементов. На этот раз команда нарастила его 17 дополнительными битами, после чего провела предобучение своей модели на 2 миллионах неразмеченных химических соединениях, взятых из датасета ZINC15. Авторы назвали такой подход самообучением графов на основе дескрипторов (Descriptor-based Graph Self-supervised Learning, DGSSL).
Чтобы приложить модель к последующей задаче (downstream task), ученые обратились к трём бенчмаркам из набора MoleculeNet: tox21 включает данные об измеренной токсичности 12 различных соединений, clintox содержит информацию о токсичности, полученную в ходе клинических испытаний FDA и toxcast содержит данные о токсичности, полученные в ходе высокопроизводительного скрининга in vitro. В роли бейзлайнов выступили результаты работы пяти современных графовых моделей, используемых для тех же целей: Infomax, JOAO, Attribute Masking, Grover, MGSSL. Серия экспериментов показала, что производительность DGSSL, выраженная через метрику ROC-AUC, на всех бенчмарках превосходит либо сопоставима с таковой у современных моделей.
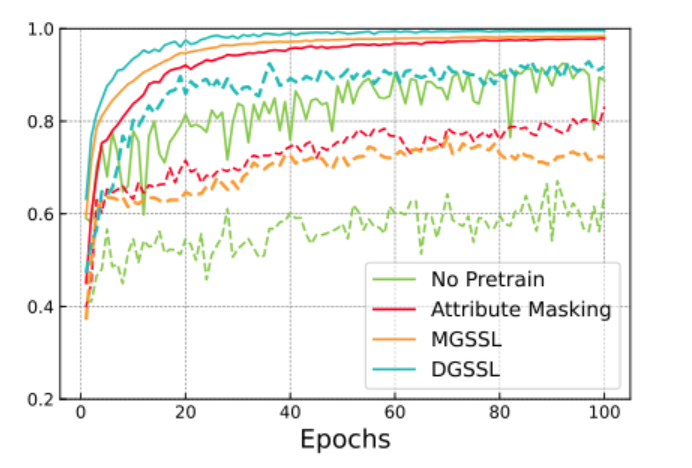
Сравнение DGSSL с другими подходами на бенчмарке clintox
Подробнее с результатами работы ученых можно познакомиться в статье, опубликованной
в журнале IEEE Access.