Константин Ушенин
Константин Ушенин
Новая нейросетевая архитектура сделала предсказание электронной плотности оптимальнее и эффективнее
Электронная плотность — это важнейший фундаментальный параметр молекул в квантовой химии. Эта функция является упрощённым способом описания многоэлектронных систем, но при этом позволяет вычислить полную энергию молекулы, её орбитали, а также множество их химических свойств.
Существует несколько подходов к расчёту электронной плотности в рамках глубокого обучения. Одним из самых популярных стал подход на основе двудольного графа, предложенный авторами модели DeepDFT. Одна часть такого графа состоит из геометрического графа, представляющего молекулу: узлам соответствуют атомы, а рёбрам — расстояния между ними.
Вторая часть состоит из точек зондирования — координат в трёхмерном пространстве в окрестностях атомов, в которых вычисляется электронная плотность. При обучении эти точки задаются с помощью равномерной трёхмерной или стандартной решёток. При инференсе же нейронной сети точки зондирования могут быть в любых позициях.
Несмотря на предложенные улучшения, архитектуры типа DeepDFT ещё далеки от оптимальных. Так, в них не учитывается большой разброс в электронной плотности (до 8 порядков), которая может окружать атомы. Это ограничивает возможности обучения DeepDFT на данных из половины самых популярных программ для химических вычислений и, как следствие, открытие таким путём новых лекарственных препаратов. Кроме того, большое количество точек на одну молекулу приводит к тому, что данные занимают очень много места и требуют много времени для обработки.
Отталкиваясь от этих проблем, исследователи из команды «Глубокое обучение в науках о жизни» AIRI предложили новую, основанную на DeepDFT архитектуру под названием LAGNet. Архитектура специально заточена для работы с похожими на лекарства молекулами (druglike molecules) — то есть соединениями, которые потенциально могут обладать биологической активностью.
Ранее мы уже рассказывали, как та же команда вместе с коллегами из других организаций собрала датасет с данными по таким молекулам ∇2DFT. Теперь же этот датасет послужил основной для обучения LAGNet. Помимо этого, исследователи применили три важные модификации к DeepDFT.
В первую очередь авторы изменили способ кодирования дистанций между атомами. Традиционно для этого используется разложение либо по базису функций Бесселя, либо по гауссинам. Новый подход объединяет несколько методов кодирования дистанций воедино, улучшая точность модели как вблизи атомов, так и на очень большом удалении от них.
Следующее улучшение, предложенное командой, заключается в сэмплировании точек с помощью стандартной решётки. Такая решётка строится заданием набора сфер вокруг каждого атома, после чего на каждой сфере точки выбираются согласно квадратурам Лебедева. Это позволило уменьшить объём данных в 8 раз по сравнению с сэмплированием по равномерной решётке. Подобные подходы хорошо зарекомендовали себя в области классических методов вычислений, но ранее в машинном обучении популярны не были.
Наконец, авторы придумали новую схему нормализации данных, названную подавлением остовных орбиталей. Её принцип основан на подборе для каждого атома коэффициентов специальной функции, которая приближает внутренние и валентные орбитали друг к другу. Приведение данных к одному масштабу существенно повысило эффективность обучения.
На основе электронной плотности, предсказанной с помощью LAGNet на равномерной сетке, можно создавать высококачественные визуализации с желаемыми изоповерхностями. Кроме того, эти данные могут быть полезны для вычисления физико-химических свойств молекул, что необходимо для поиска новых лекарств.
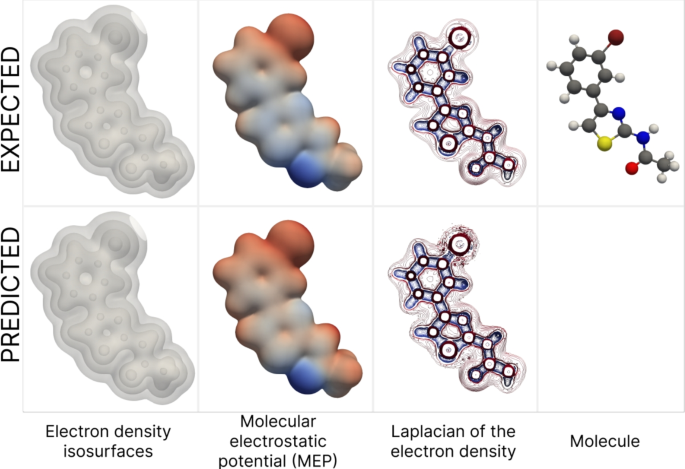
Визуализация различных свойств молекулы, взятых из датасета ∇2DFT, и предсказанных LAGNet
Исследование опубликовано в журнале Journal of Cheminformatics.