Марат Хамадеев
Марат Хамадеев
Исследователи усовершенствовали модель на основе обучения с подкреплением для поиска новых лекарств
Лекарственные вещества интересовали людей ещё в античности. С тех пор фармакология развилась в целую науку. Но, несмотря на значительный прогресс, для многих заболеваний всё ещё нет лекарственной терапии. По этой причине создание новых препаратов продолжает быть актуальной задачей.
Долгое время единственным способом найти новое лекарство оставались эксперименты в лаборатории. С развитием вычислительной техники в XX веке появились численные методы молекулярного моделирования, что позволило уменьшить количество рассматриваемых кандидатов на этапе in vitro и в целом сократить весь процесс поиска. В случае с лекарствами необходимо найти такую молекулу, которая при связывании с белком-мишенью существенно изменит его функциональное состояние. Под белком-мишенью в данном случае понимается молекула, вовлеченная в конкретный метаболический или сигнальный путь, связанный с определенным болезненным состоянием.
Традиционно такой поиск делается с помощью различных численных методов. Например, во время виртуального скрининга алгоритм отбирает подходящие химические соединения из базы данных, основываясь на численных оценках характеристик взаимодействий белка и лиганда, полученных молекулярным моделированием. Однако подобные базы не охватывают все возможные молекулы, что исключает подбор ранее неизвестных соединений. Кроме того, перебор всех вариантов зачастую может быть слишком дорогим.
В последние годы приобретают популярность генеративные подходы, основанные на глубоком обучении, которые способны обойти эти ограничения. К ним относятся подходы, основанные на обучении с подкреплением (Reinforcement Learning, RL). В них пространство состояний, в котором идёт поиск, представляется с помощью множества всевозможных молекулярных графов, в узлах которого располагаются атомы, а рёбра соответствуют химическим связям. Присоединение же или удаления атома или молекулярного фрагмента можно рассматривать как действие агента.
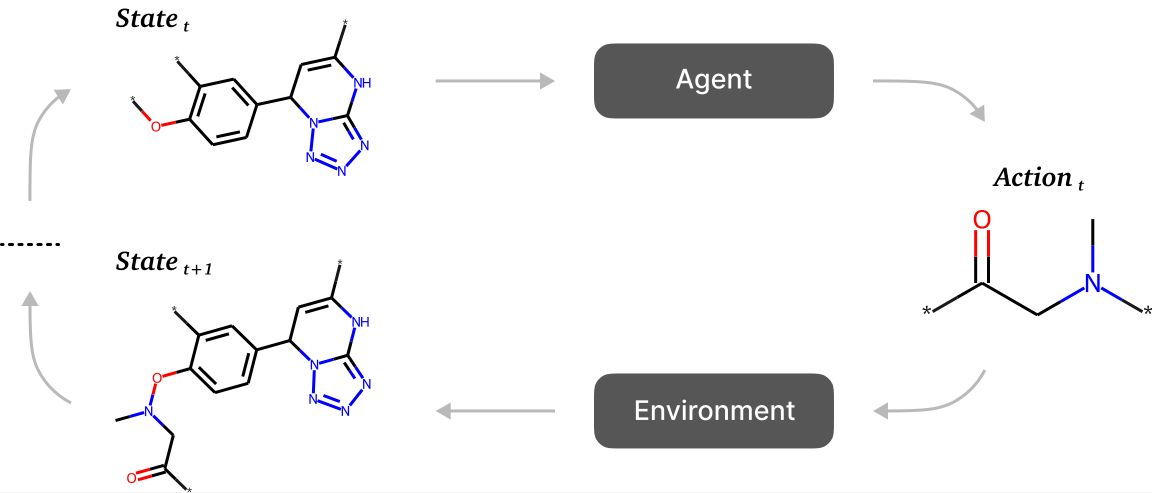
Схема RL-подхода к генерации молекул
Одним из самых многообещающих RL-подходов к поиску нужных молекул стал FREED, представленный в 2021 году. FREED решает описанную задачу, оптимизируя оценку энергии связывания лиганда с белком.
Исследователи из команды «Глубокое обучение в науках о жизни» AIRI проявили к FREED большой интерес. Они, однако, обнаружили, что эта модель содержит баги и неэффективные блоки, а также обладает ещё целым рядом недостатков.
Сначала учёные исправили все баги и неконсистентности, что увеличило качество генерации. Получившаяся модель получила название FFREED (Fixed FREED). На следующем шаге исследователи упростили архитектуру актора, что в конечном итоге ускорило поиск. Финальная модель, названная FREED++, оказалась быстрее в 8,5 раза и в 22 раза менее затратной по памяти, чем модель FFREED. Кроме того, ускорить и уменьшить модель получилось без каких-либо потерь в качестве генерации. Исследователи показали, что модели FFREED и FREED++ способны генерировать молекулы с более высокими значениями аффинности, чем существующие подходы.
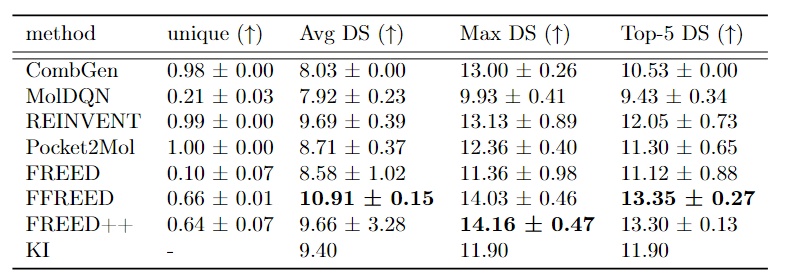
Таблица сравнения FFREED и FREED++ с альтернативными подходами, включая FREED на задаче оптимизации аффинности лиганда к белку USP7. В таблице представлены процент уникальных сгенерированных молекул, а также характеристики распределения значений энергии связывания с белком.
Статья с техническими подробностями опубликована в журнале Transactions on Machine Learning Research, код доступен на GitHub.