Марат Хамадеев
Марат Хамадеев
Новый фреймворк потребовал в 50 раз меньше данных для предсказания оптимальной геометрии молекул с помощью нейросетей
Чтобы создать новое лекарство или материал, учёные должны исследовать молекулы, которые ещё никто не синтезировал, и надеяться найти у них полезные свойства. В XX веке эта поисковая работа велась в основном экспериментально, но в наши дни такой подход оказывается слишком затратным.
Вместо этого специалисты обращаются за помощью к компьютерному моделированию. Среди физических симуляторов, решающих такие задачи, популярностью пользуются методы на основе функционала плотности (density functional theory, DFT). Они позволяют с необходимой точностью предсказывать энергии молекулярных конформаций. При этом наиболее ценным является поиск таких геометрий молекулы, в которых достигается локальный минимум энергии, так как это наиболее вероятные конфигурации, в которых молекула вступает в химическую реакцию.Такой подход существенно быстрее «мокрого» эксперимента по синтезу и дальнейшей характеризации молекулы, но всё же достаточно долог: одна итерация этого метода для большой молекулы может требовать нескольких часов вычислений.
По этой причине в последние годы активно развиваются подходы, которые используют нейронные сети для предсказания молекулярных свойств за более короткое время. Работа в этом направлении включает в себя подготовку датасетов, одним из которых стал датасет nablaDFT, собранный учеными Института AIRI, Сколтеха и ПОМИ РАН.
Существует несколько способов применить глубокое обучение к задачам квантовой химии. Например, часть исследователей рассматривает предсказание оптимальной геометрии как генеративную задачу. Другой подход основан на нейросетевом выучивании потенциалов (Neural Network Potentials, NNP) в которых находятся атомы, что позволяет вычислять потенциальную энергию молекулярной конформации и строить силовые поля (molecular force field, MFF). Такая техника сводит задачу к градиентной оптимизации без необходимости задействовать физический симулятор, что существенно сокращает вычислительную сложность.
Команда исследователей из AIRI, ФИЦ ИУ РАН, МФТИ и Университета Констрактор в Бремене протестировала NNP подход на части датасета nablaDFT. Она обнаружила, что конформационная оптимизация систематически страдает от смещения (distribution shift), что приводит к неточной минимизации энергии.
Для борьбы с этой проблемой учёные предложили обогащать обучающую выборку оптимизационными траекториями. Под такой траекторией понимается последовательность конформаций, рассчитанных с помощью физического симулятора, которая в пространстве параметров (координаты и типы атомов) стремится к оптимальному решению. Оказалось, что таким образом действительно можно нивелировать смещение, правда, стоимость таких вычислений велика. Так, расчёт дополнительных 500 тысяч конформаций потребовал 9 CPU-лет вычислений.
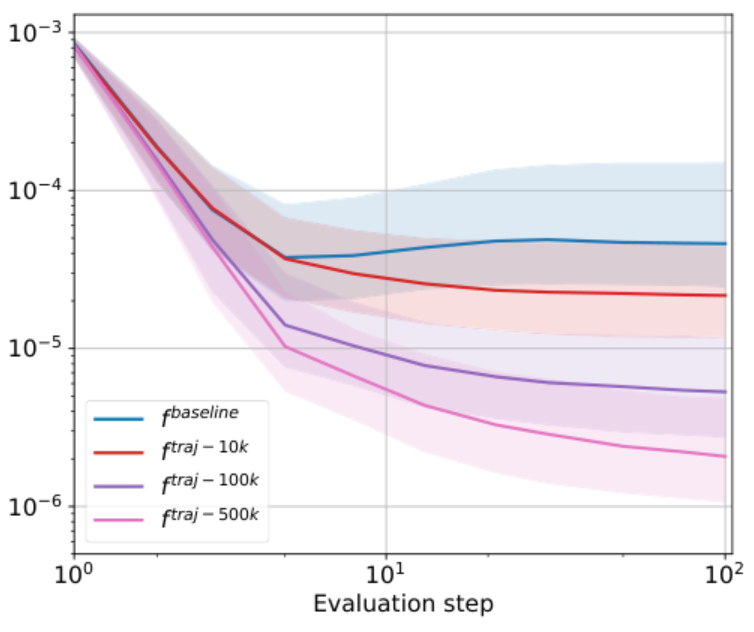
Зависимость ошибки предсказания межатомных сил от шага оптимизации для нейронных потенциалов с разным числом дополнительных конформаций
Пытаясь снизить количество требуемых данных, авторы предложили использовать активное обучение. В его основе лежит отбор и добавление в выборку только тех конформаций, на которых модель ошибается. В роли инструмента, который проводит такой отбор, исследователи использовали дешёвый и быстрый MFF-оракул. Выбранные с его помощью данные обсчитываются с помощью DFT-оракула и добавляются в обучающую выборку. Поскольку обновление выборки в таком подходе происходит поэтапно, авторы назвали его постепенным выучиванием оптимизации (Gradual Optimization Learning Framework, GOLF).
Для оценки качества нового подхода исследователи сравнили его с работой нейронных потенциалов, дообученных на разном количестве данных из nablaDFT, а также с некоторыми генеративными подходами (Torsional Diffusion, ConfOpt, Uni-Mol+). В качестве критериев оценки ими были выбраны метрики, основанные на энергии. Проверка показала, что нейронный потенциал, обученный с помощью GOLF на 10000 конформаций, имеет такую же ошибку, как и базовая NNP-модель, обученная на 500000 дополнительных конформаций.
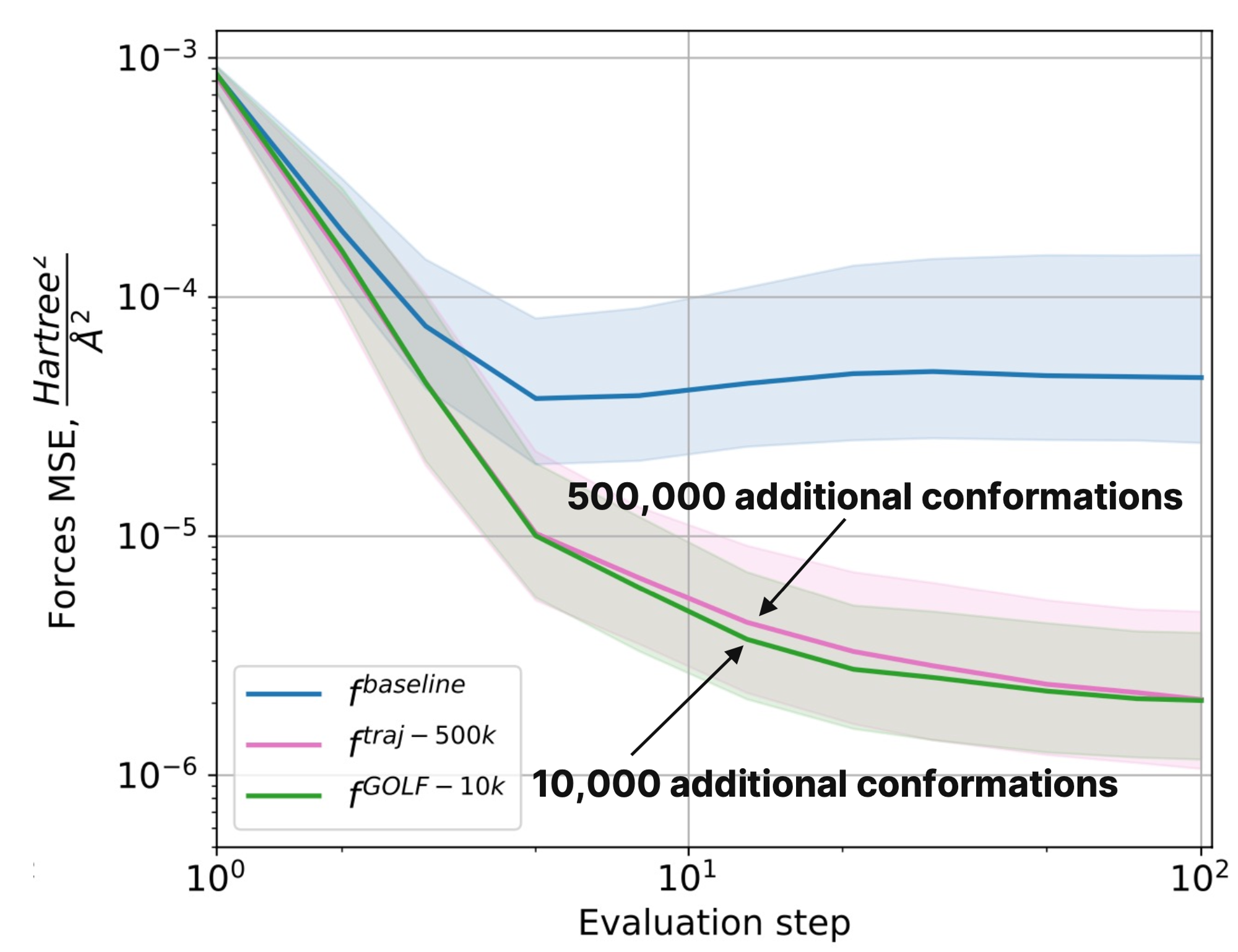
Зависимость ошибки предсказания межатомных сил от шага оптимизации для нейронных потенциалов
Авторы надеются, что новый подход будет особенно полезен в тех случаях, когда собрать большое количество оптимизационных траекторий может быть затруднительно. К ним относятся такие сложные атомные системы, как адсорбент-адсорбат, молекула в растворе, молекула в белке и так далее.
Подробности исследования можно найти статье, опубликованной в сборнике конференции ICLR 2024, код доступен на GitHub.