Марат Хамадеев
Марат Хамадеев
Новый датасет с молекулярными данными поможет обучать и проверять нейросети для предсказания химических свойств
Предсказание свойств новых соединений на компьютере облегчило часть работы по проверке гипотез в химии. Сегодня численные методы обрели новое дыхание благодаря нейросетям. Используя способность последних к аппроксимации, можно напрямую вычислять нужное свойство, отталкиваясь от химической формулы. Мы уже рассказывали, как для этого используют трансформеры и графовые нейронные сети.
Другой класс подходов использует методы машинного обучения для сокращения вычислений в рамках квантовой химии. В этом случае целью алгоритма может стать расчёт многоэлектронной волновой функции, функции электронной плотности для метода DFT или матрицы DFT-гамильтониана. Знание этих физических величин позволяет вычислять нужные свойства из первых принципов (ab initio).
Каждый из подходов этого класса имеет свои преимущества и недостатки. Так, аппроксимация матрицы гамильтониана обладает удовлетворительной точностью и ускоряет сходимость DFT-алгоритмов, но хорошо работает только в рамках одной молекулярной формулы. Это ограничение можно было бы обойти с помощью больших датасетов.
Один из таких датасетов собрала команда исследователей из AIRI, Сколтеха, МФТИ и ПОМИ РАН в 2022 году. Получивший название ∇DFT набор данных содержит информацию о более, чем миллионе молекул и пяти миллионах конформаций.
Недавно же большой коллектив учёных из групп «Глубокое обучение в науках о жизни» и «Прикладное NLP» AIRI и их коллег из EPFL, СПбГУ, ИСП РАН и ПОМИ РАН выпустил его следующую версию под названием ∇2DFT, увеличив количество данных до почти двух миллионов молекул, а количество конформаций — до 15,7 миллиона. На его основе авторы сделали бенчмарк из трех типов задач: предсказание матриц гамильтониана, предсказание энергии и атомных сил и оптимизация геометрии молекулы.
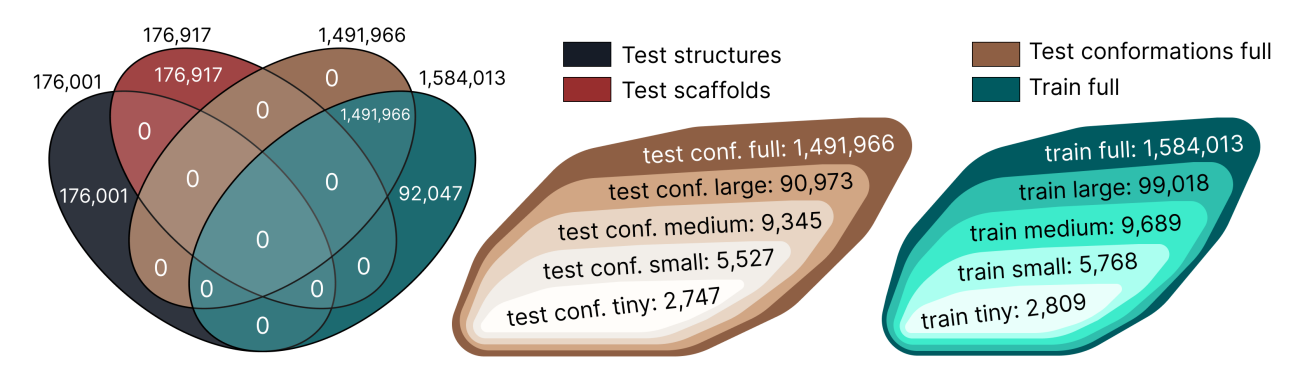
Структура ∇2DFT. Датасет был вручную разбит на тестовую и обучающую выборки 12 разными способами, чтобы обеспечить гибкость при выборе дизайна экспериментов.
Исследователи разместили базу данных общим объёмом 220 терабайт в открытый доступ на платформу Cloud. Помимо молекулярных данных в него входят 10 моделей для предсказания энергии и атомных сил в конформациях и 3 модели для работы с теорией функционала плотности.
Подробнее о новом датасете можно прочитать в статье, опубликованной в сборнике трудов конференции NeurIPS 2024, примеры работы с ним и код выложены на GitHub.