Marat Khamadeev
Marat Khamadeev
The new framework required 50 times less data to predict the optimal geometry of molecules using neural networks
To create a new drug or material, scientists must research molecules that have yet to be synthesized and hope to find valuable properties. In the 20th century, this search was mainly conducted experimentally, but nowadays, such an approach is too costly.
Instead, experts turn to computer modeling for assistance. Among the physical simulators used to solve such problems, methods based on density functional theory (DFT) are popular. They allow for the prediction of the energies of molecular conformations with the necessary precision. An important task is to find molecular geometries where a local energy minimum is achieved, as these are the most likely configurations in which the molecule undergoes a chemical reaction. This approach is significantly faster than “wet” experiments involving the synthesis and further characterization of the molecule. However, it is still quite time-consuming: one iteration of this method for a large molecule may require several CPU hours.
Recently, neural networks have significantly reduced the time required to predict molecular properties. Training such neural networks involves preparing specific datasets. One of these datasets is the nablaDFT, compiled by scientists from the AIRI Institute, Skoltech, and PDMI RAS.
There are several ways to apply deep learning to finding low-energy conformations. For example, one approach is to reformulate this task as a conditional generation task. Another approach is to train neural network potentials (NNP) to predict the potential energy of a molecular conformation and use it as a molecular force field (MFF) for relaxation. Such a technique allows for gradient-based optimization without a physical simulator, significantly reducing computational complexity.
A team of researchers from AIRI, FRC CSC RAS, MIPT, and the Constructor University tested the NNP approach on the part of the nablaDFT dataset. They have found that conformational optimization systematically suffers from distribution shift, leading to inaccurate energy minimization.
Scientists proposed enriching the training set with optimization trajectories to address this issue. An optimization trajectory is a sequence of conformations calculated using a physical simulator that aims for an optimal solution in parameter space (coordinates and types of atoms). It was found that this approach indeed mitigates the shift; however, the cost of such computations is high. For instance, calculating an additional 500,000 conformations required 9 CPU-years of computation.
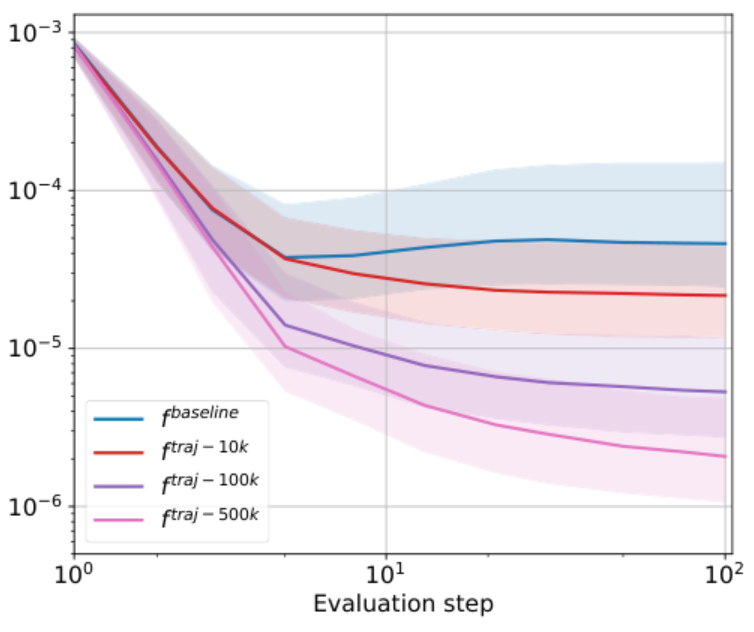
Prediction error of interatomic forces vs the optimization step for neural potentials with different numbers of additional conformations
In an attempt to reduce the amount of required data, the authors proposed using active learning. The proposed approach reduces the number of conformations added to the training set by only selecting those conformations where the model’s prediction is incorrect. Researchers used a cheap and fast MFF oracle as a tool for such selection. The selected data, calculated using the DFT oracle, is added to the training set. Since the training set is updated gradually in this approach, the authors named it the Gradual Optimization Learning Framework (GOLF).
To evaluate the quality of the new approach, researchers compared it with Neural Network Potentials retrained on different amounts of data from nablaDFT and with some generative approaches (Torsional Diffusion, ConfOpt, Uni-Mol+). Energy-based metrics were chosen as evaluation criteria. The evaluation showed that a neural potential trained using GOLF on 10,000 conformations has the same error as the baseline NNP model trained on 500,000 additional conformations.
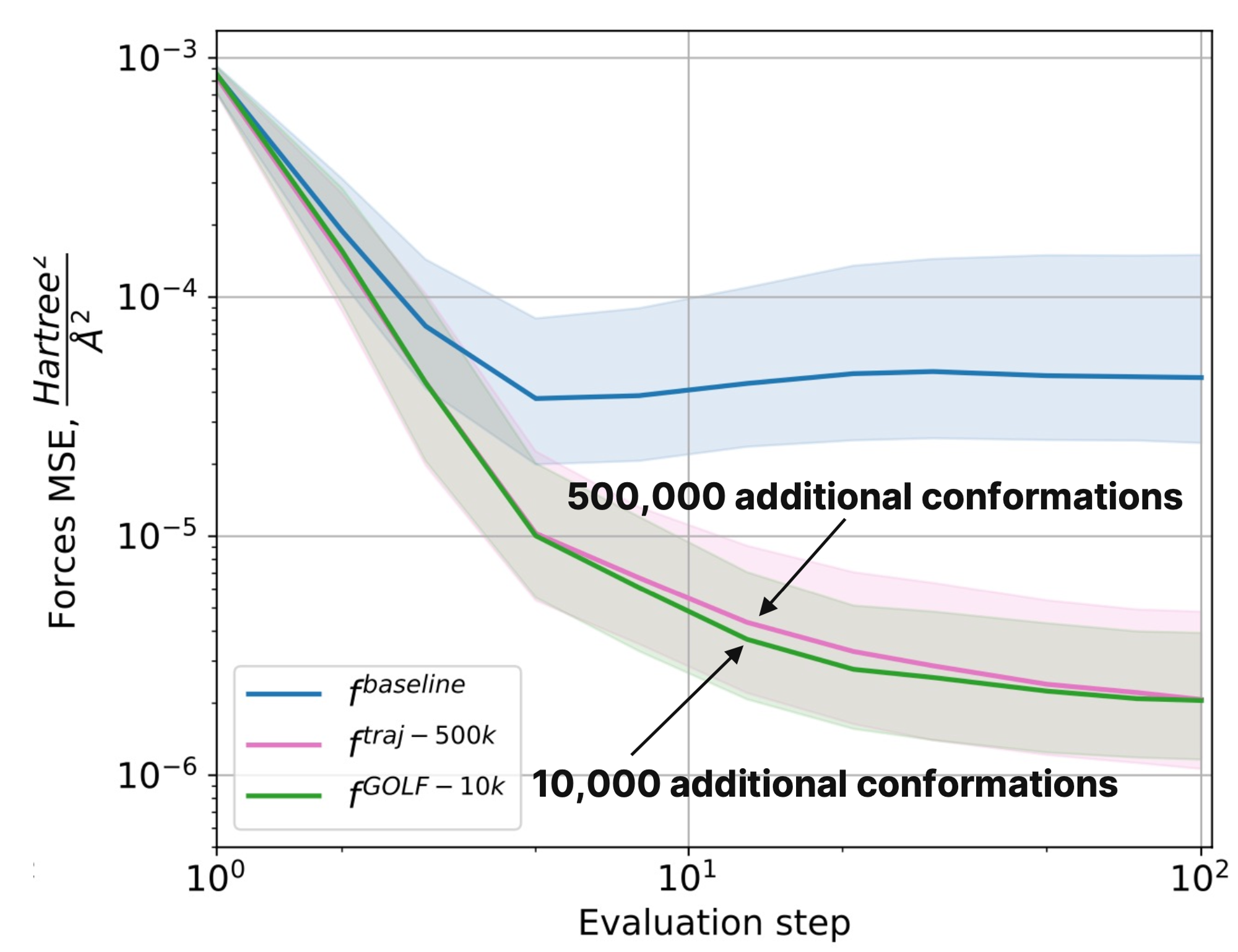
Prediction error of interatomic forces vs. the optimization step for neural potentials
The authors hope that the new approach will be particularly useful in cases where collecting a large number of optimization trajectories can be challenging. These include complex atomic systems such as adsorbent-adsorbate, solution, protein complexes, and so on.
Further details of the study can be found in the paper published in the ICLR 2024 conference proceedings, and the code is available on GitHub.