Marat Khamadeev
Marat Khamadeev
A new dataset with molecular data will help to train and test neural networks predicting chemical properties
The prediction of properties of new compounds on a computer has simplified part of the work involved in testing hypotheses in chemistry. Today, numerical methods have gained new momentum thanks to neural networks. By utilizing the ability of the latter to approximate, it is possible to directly compute the desired property based on the chemical formula. We have previously discussed how transformers and graph neural networks are used for this purpose.
Another class of approaches employs machine learning methods to reduce computations within the framework of quantum chemistry. In this case, the algorithm's goal may be to calculate the multi-electron wave function, the electron density function for the DFT method, or the DFT Hamiltonian matrix. Knowledge of these physical quantities allows for the computation of necessary properties ab initio.
Each approach in this class has its advantages and disadvantages. For instance, approximation of the Hamiltonian matrix offers good accuracy and accelerates the convergence of DFT algorithms, but it works well only within a single molecular formula. This limitation could be overcome with large datasets.
One such dataset was assembled by a team of researchers from AIRI, Skoltech, MIPT and PDMI RAS in 2022. The ∇DFT dataset contains information about more than one million molecules and five million conformations.
Recently, the large collaborations of scientists from the “DL in Life Sciences” and “Domain-specific NLP” groups of AIRI and their colleagues from EPFL, St. Petersburg State University, ISP RAS, and PDMI RAS, released its next version, ∇2DFT, increasing the number of data points to nearly two million molecules and the number of conformations to 15.7 million. Based on this, the authors created a benchmark consisting of three tasks: predicting Hamiltonian matrices, predicting energy and atomic forces, and optimizing molecular geometry.
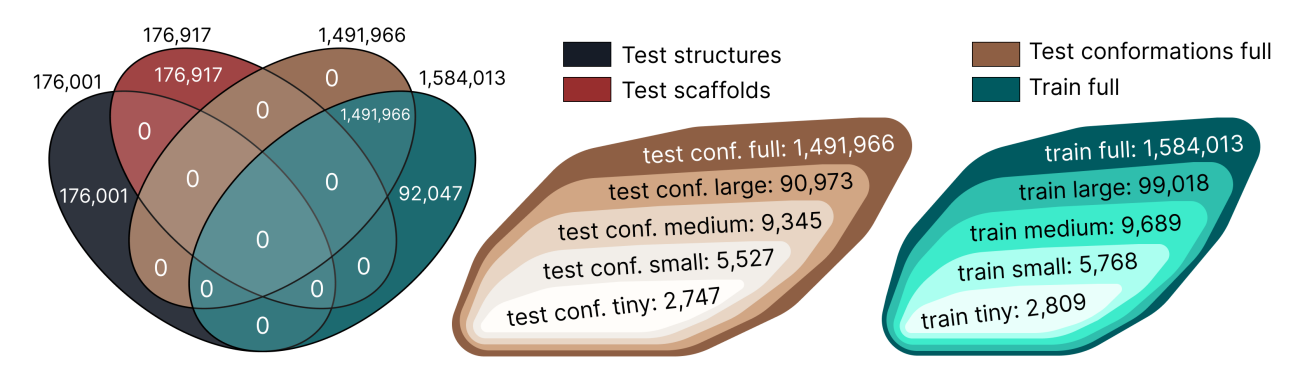
Structure of ∇2DFT. The dataset was manually divided into test and training samples in 12 different ways to ensure flexibility in experimental design.
The researchers made the database, totaling 220 terabytes, publicly available on the Cloud platform. In addition to molecular data, it includes 10 models for predicting energy and atomic forces in conformations and 3 models for working with DFT.
More details about the new dataset can be found in an article published in the proceedings of the NeurIPS 2024 conference, with examples of working with it and code available on GitHub.