Marat Khamadeev
Marat Khamadeev
Descriptor-Based Graph Self-Supervised Learning helped to predict molecule toxicity
More than a hundred chemical elements are known today and arranged in the periodic table. These elements can combine into molecules, the variety of which is countless. Each molecule is characterized by its elemental composition and structure, which, according to complex quantum mechanical rules, determine its biochemical properties. Understanding these properties is important for many areas of human activity, primarily for the search for new drugs.
The traditional approach to discovering these properties is through laboratory experiments with solutions which is why it is called "wet lab". In contrast, molecular biology has actively developed “dry” computational methods over the past half-century, in which scientists predict the properties of chemical compounds using computers.
During this time, computational biology methods have evolved significantly, and today researchers are pinning their hopes on deep learning. Graph neural networks have proven to be effective in predicting the properties of molecules, as molecules can be naturally represented as a graph: atoms are located at their nodes, and edges correspond to chemical bonds.
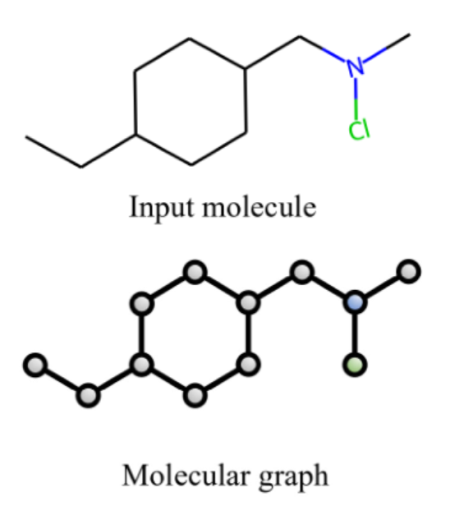
Representing a molecule using a graph
To begin deep learning, a large amount of labeled data is usually required: information about the molecule's structure, its constituent atoms and bonds, and the specific values of its properties, such as toxicity, obtained through "wet" experiments. However, there is much more unlabeled data than labeled data, and collecting additional labeling can be expensive. Therefore, self-supervised learning (SSL) is becoming popular, in which internal properties of the data are used as labels.
This technique starts with pre-training on unlabeled data, the task of which is to independently learn the representation of objects, after which the model can be used in supervised learning mode on a new small amount of labeled data. The known strategies for self-supervised learning of graph neural networks applied to molecules either use node-level pre-training involving masking and predicting atomic properties or capture rich information in subgraphs with motif-based methods.
A group of scientists from HSE, AIRI and NUST MISIS led by Ilya Makarov is confident that these approaches are not optimal, as node-level auxiliary tasks do not preserve useful domain knowledge, and the fusion of motif-based methods and node-level tasks is computationally extensive. Instead, the team developed an approach to self-supervised learning of graphs based on molecular descriptors.
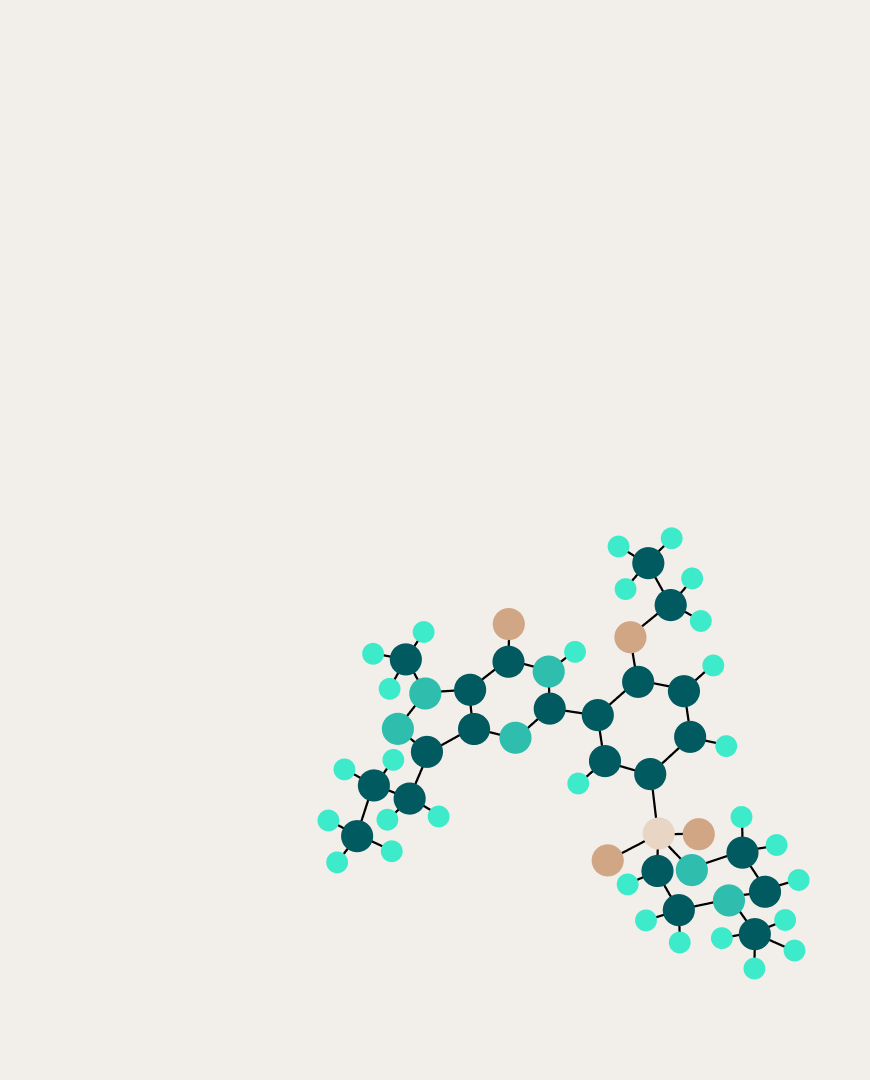
Molecular descriptors are certain numerical values that express chemical properties through elemental composition and structure. A molecule can be divided into substructures containing one or more atoms according to their biological significance — in this case, by the presence of electrons forming a π-bond. A special descriptor language for describing such centers is known as the fragmentary code of substructure superposition (FCSS). Descriptor centers can consist of heteroatoms, carbons connected by double or triple bonds, and aromatic systems. Since aromaticity can be determined by the properties of atoms or bonds (if the atom or bond is aromatic), the authors focused only on the first two cases and gathered 17 types of descriptor center patterns from FCSS.
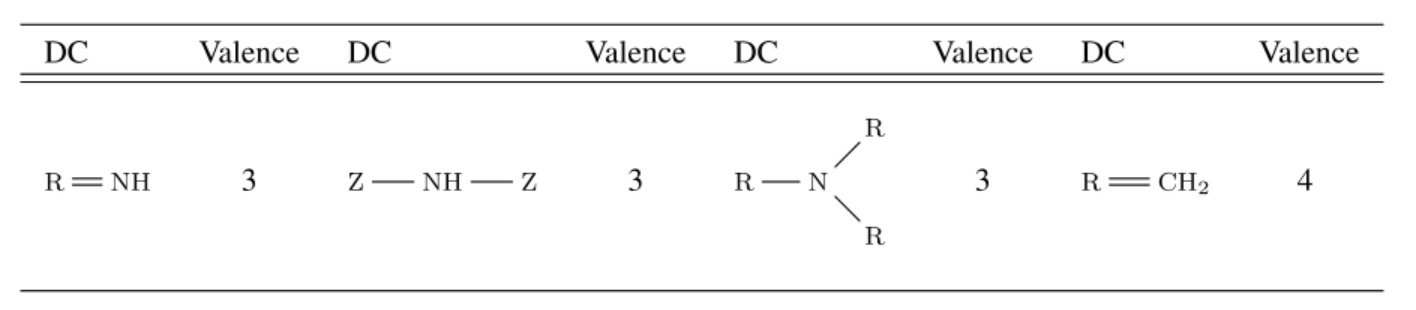
List of 4 out of the 17 selected FCSS descriptor centers (Z represents any atom, R represents any atom except for H)
The researchers designed a node-level prediction task that integrates domain knowledge from descriptor centers, wh ere the semantics of local subgraphs are encoded into the prediction target. Typically, it is solved based on a vector defined to encode 119 atomic symbols. This time, the team added to the vector 17 descriptor centers as special atomic numbers and pre-trained their model using a dataset of 2 million unlabeled compounds sampled from the ZINC15 dataset. The authors named this approach Descriptor-based Graph Self-supervised Learning (DGSSL).
To apply the model to a downstream task, the scientists selected three toxicity-related benchmarks from the MoleculeNet dataset: tox21 comprises toxicity measurements for 12 biological targets, clintox includes toxicity information from FDA clinical trials, and toxcast contains toxicity data obtained through in vitro high-throughput screening. The results of five modern graph models used for the same purposes — Infomax, JOAO, Attribute Masking, Grover, and MGSSL — served as baselines. A series of experiments showed that the performance of DGSSL, expressed through the ROC-AUC metric, outperforms or is comparable to that of modern models on all benchmarks.
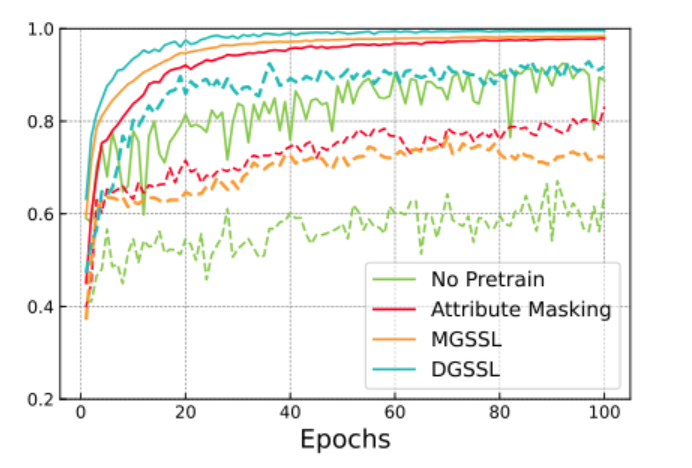
Comparison of DGSSL with other approaches on the clintox benchmark. Solid and dashed lines indicate training and testing curves, respectively
More details of the study can be found in an article published in IEEE Access journal.