Konstantin Ushenin
Konstantin Ushenin
A new neural network architecture will make electron density prediction better
Electron density is one of the most fundamental parameters in quantum chemistry. It provides a simplified yet powerful way to describe many-electron systems, allowing researchers to compute a molecule’s total energy, molecular orbitals and a wide range of chemical properties.
Several deep learning approaches have been proposed to predict electron density, with one of the most popular being a bipartite graph-based method introduced by the authors of the DeepDFT model. In this approach, one part of the graph represents the molecular geometry: atoms are nodes, and edges encode distances between atoms. The second part consists of probe points — 3D coordinates in the vicinity of atoms where the electron density is evaluated. During training, these points are arranged in uniform 3D or the standart grids. At inference time, however, the model can predict density at arbitrary positions in space.
Despite several advancements, architectures like DeepDFT are still far from optimal. For instance, they struggle with the wide dynamic range of electron density values (up to 8 orders of magnitude), which poses a serious challenge for machine learning models. This limits training of DeepDFT with data from half of the most popular chemical software and, hence, limits applications related to drug discovery. Moreover, the high number of sampling points per molecule leads to massive datasets that are expensive to store and process.
To address these issues, researchers from the DL in Life Sciences team at AIRI have proposed a new architecture, LAGNet, based on DeepDFT but tailored for druglike molecules — compounds with potential biological activity.
The team had previously collaborated with other institutions to create a dataset of such molecules, known as ∇2DFT, which now serves as the foundation for training LAGNet. In addition, the researchers introduced three major modifications to the DeepDFT architecture.
First of all, the authors changed the way the distances between atoms are encoded. Instead of relying on traditional encodings such as Bessel function or Gaussian basis expansions, the team developed a method that combines multiple distance encoding strategies. This hybrid approach improves prediction accuracy both near atomic cores and in regions far from them.
Next, the researchers proposed a new sampling strategy based on a standard grid. Around each atom, a set of concentric spheres is defined, and points on each sphere are selected using Lebedev quadrature. This method reduced data volume by a factor of 8 compared to uniform grids. Such approaches are well-established in the field of computation methods, but were not famous in machine learning before.
Finally, the authors introduced a novel scheme for data normalization that is called a core suppression. For each atom, they fit a custom scaling function that brings core and valence orbitals closer together in magnitude. This normalization significantly improves learning efficiency by reducing the dynamic range of the target values.
Based on the predicted electron density on a uniform grid, LAGNet enables the generation of high-quality visualizations with customizable isosurfaces. These predictions also serve as a valuable input for calculating physicochemical properties of molecules — an essential step in modern drug discovery.
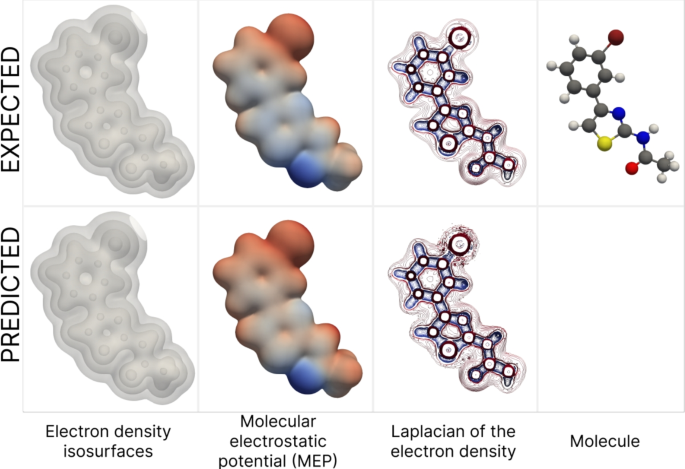
Examples of downstream tasks with target and predicted electron density for a molecule from the ∇2DFT dataset
The study was published in the Journal of Cheminformatics.