Artem Dembitskiy
Artem Dembitskiy
Machine learning helped to discover promising lithium-ion conductors
Lithium-ion conductors play a crucial role in a wide range of electrochemical applications, with lithium-ion batteries being the most prominent example. Enhancing the performance of these devices can be achieved by selecting materials with improved properties. For instance, higher lithium-ion conductivity can lead to faster charging and increased battery power.
Finding new materials, however, remains a nontrivial and complex task. Traditionally, researchers have relied heavily on chemical intuition, synthesizing candidate substances in the lab and testing their properties experimentally. Today, this part of the process can be made faster using numerical simulations, though even theoretical methods can be time-consuming.
In recent years, faster computational approaches based on machine learning have been gaining traction. In the tasks of ion transport, machine learning interatomic potentials (MLIPs) have shown great promise, allowing the estimation of a chemical system's energy based on atomic positions.
With sufficiently large volumes of quantum chemistry data collected for various structures and chemical compositions, it becomes possible to train universal MLIPs (uMLIPs). These models are expected to generalize to compounds not seen during training. However, until now, there has been no comparative framework to assess the reliability and accuracy of uMLIPs in predicting the transport properties of lithium-ion conductors.
A research team from Skoltech and AIRI addressed this issue by compiling a large dataset calculated using density functional theory (DFT). The dataset, named LiTraj, contains 13,000 percolation thresholds and 122,000 lithium-ion migration barriers across various crystal structures encompassing 45 chemical elements. In addition, it includes 1,700 initial and optimized migration trajectories obtained through nearly one million optimization iterations.
Using this dataset, the authors compared the accuracy of several existing uMLIPs — M3GNet, CHGNet, MACE-MP, and SevenNet — in optimizing lithium-ion migration trajectories. They found that two models stood out in terms of performance: SevenNet and MACE-MP. The researchers fine-tuned these models on their dataset, achieving a mean absolute error of just 0.1 eV in predicting migration barriers — an accuracy sufficient for large-scale lithium-ion conductor screening.
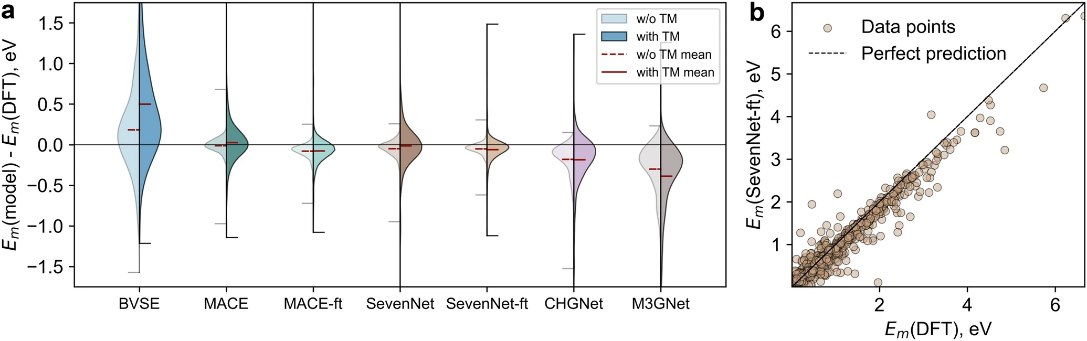
Results of the comparative analysis of several uMLIPs. Left — the prediction error distributions for each model for different subsets of the dataset; the “ft” suffix indicates fine-tuned models. Right — the parity plot for the predictions of the SevenNet-ft uMLIP.
The researchers also demonstrated the utility of uMLIPs in searching for protective cathode coating materials in solid-state batteries. In the early stages, they filtered out compounds with undesirable elemental compositions (e.g., expensive, toxic, or radioactive elements). The list was then further narrowed based on other criteria such as thermodynamic stability and compatibility with electrolyte and cathode materials.
Ultimately, the initial set of 13,000 lithium-containing compounds was reduced to around 100 structures that met all criteria. Ion mobility calculations using the fine-tuned uMLIP revealed several promising candidates, including Li₃AlF₆, LiYF₄, Li₃PO₄, and Li₂ZnCl₄.
The research article is published in npj Computational Materials, and the dataset is openly available on GitHub.