Marat Khamadeev
Marat Khamadeev
Researchers improved a reinforcement learning-based model for drug discovery
Medications have intrigued people since ancient times. Since then, pharmacology has evolved into a full-fledged science. However, despite significant progress, there is still no drug therapy for many diseases. For this reason, the design of new drugs remains a relevant task.
For a long time, the only way to discover a new drug was through laboratory experiments. With the development of computing in the 20th century, numerical methods of molecular modeling emerged, allowing researchers to reduce the number of candidates considered at the in vitro stage and, in general, to shorten the entire drug discovery cycle. In the case of drugs, it is necessary to find a molecular structure with desired biological functionality, e.g., an ability to activate or suppress a target protein via binding to it. Here, the target protein refers to a molecule involved in a specific metabolic or signaling pathway associated with a particular disease state.
Traditionally, this search has been conducted using various numerical methods. For example, during virtual screening, an algorithm selects suitable chemical compounds from a database based on an estimation of protein-ligand interaction characteristics obtained by molecular modeling. However, such databases do not cover all possible molecules, which excludes the identification of previously unknown compounds. Additionally, exploring all options can often be too expensive.
In recent years, generative approaches based on deep learning have gained popularity, capable of overcoming these limitations. These include approaches based on reinforcement learning (RL). In these methods, the state space in which the search occurs is represented by a set of various molecular graphs, where atoms represent graph nodes and edges correspond to chemical bonds. The addition or removal of an atom or molecular fragment can be viewed as an action taken by the agent.
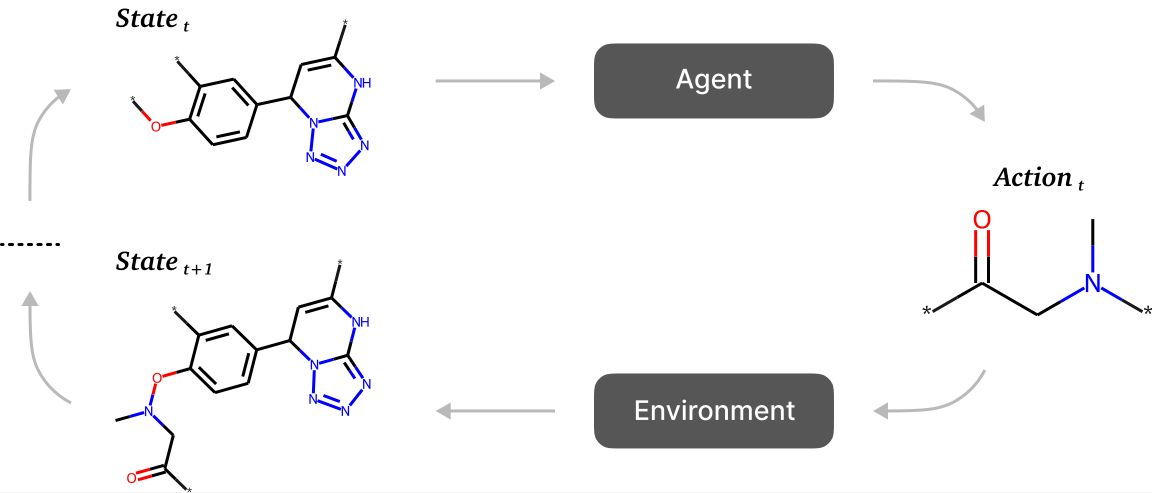
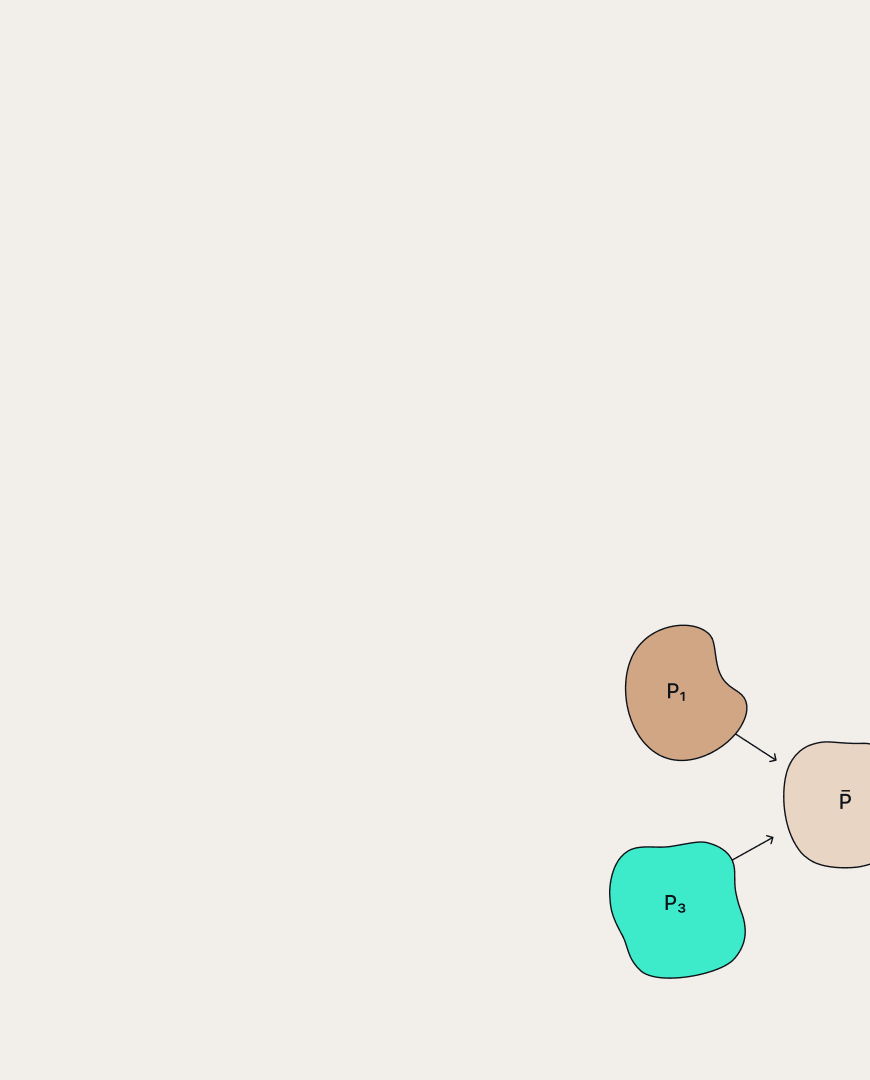
A scheme of RL-based sequential generation methods
One of the most promising RL approaches for finding the desired molecules is FREED presented in 2021. FREED solves the task by optimizing the estimate of protein-ligand binding affinity.
Researchers from the "DL in Life Sciences" team at AIRI strongly believe in the approach used in FREED. However, they found that this model contains bugs and inefficient blocks, and also has several other limitations.
Initially, the scientists fixed all the bugs and inconsistencies, which improved the quality of generation. The resulting model was dubbed FFREED (Fixed FREED). In the next step, the researchers simplified the actor architecture, which ultimately accelerated the search. The final model, named FREED++, turned out to be 8.5 times faster and 22 times less memory-intensive than the FFREED model. Additionally, the model was accelerated and simplified without any loss in generation quality. The authors demonstrated that both FFREED and FREED++ models are capable of generating molecules with higher affinity values than existing approaches.
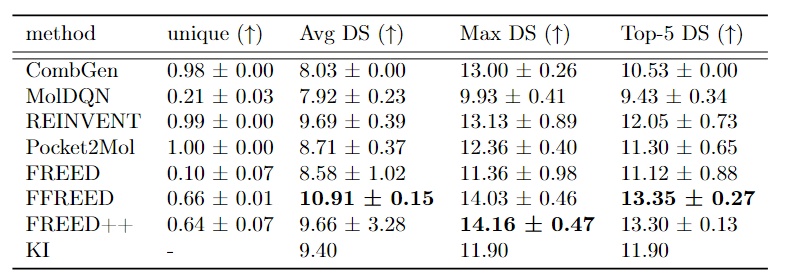
Comparison of FREED, FFREED and FREED++ with baselines on docking score optimization task for USP7 target. The table presents the percentage of unique generated molecules as well as characteristics of the distribution of binding affinity.
The article with technical details has been published in the journal Transactions on Machine Learning Research, and the code is available on GitHub.