Marat Khamadeev
Marat Khamadeev
AlphaFold2 has been modified to better assess the effects of amino acid point mutations on protein structure
Proteins play a crucial role in living organisms, performing many key functions and sustaining the very processes of life. How a protein acts and what properties it exhibits depend not only on its amino acid sequence but also on the structure into which the protein folds during the folding process.
Calculating the folded structure of a protein from first principles is virtually impossible. At the same time, biochemistry recognizes Anfinsen’s dogma, which states that the three-dimensional structure of a protein in its standard physiological environment is determined solely by its amino acid sequence. While not an absolute rule, it holds for most proteins. This principle has paved the way for the use of machine learning to predict protein structures and even design proteins — selecting ones with desired properties and functions.
One of the most impressive achievements in this area is the creation of AlphaFold2, whose developers were awarded the 2024 Nobel Prize in Chemistry. The model has proven useful not only for predicting three-dimensional structures but also for solving a range of other protein design tasks: creating new proteins without relying on existing ones, iteratively optimizing amino acid sequences, evaluating the designability of candidates, and more.
However, when assessing the impact of mutations on protein stability, AlphaFold2 demonstrates limited accuracy. This is largely due to its repeated reuse (recycling) of structural information from the non-mutated (wildtype) protein template. As a result, the mutated structures predicted by the model differ little from the originals, preventing it from fully capturing the effects of single mutations.
This limitation motivated a research team from AIRI to enhance the AlphaFold2 pipeline, leading to the development of the AFToolkit framework. Its foundation lies in several modifications, the key one being a restriction on template use in recycling stages. Importantly, this modification affects only AlphaFold2 inference and does not require retraining the model. Using AlphaFold2 embeddings in combination with lightweight adapters allowed the researchers to achieve better performance across various mutation-related tasks, such as prediction of protein stability change, protein–protein binding affinity change, and others.
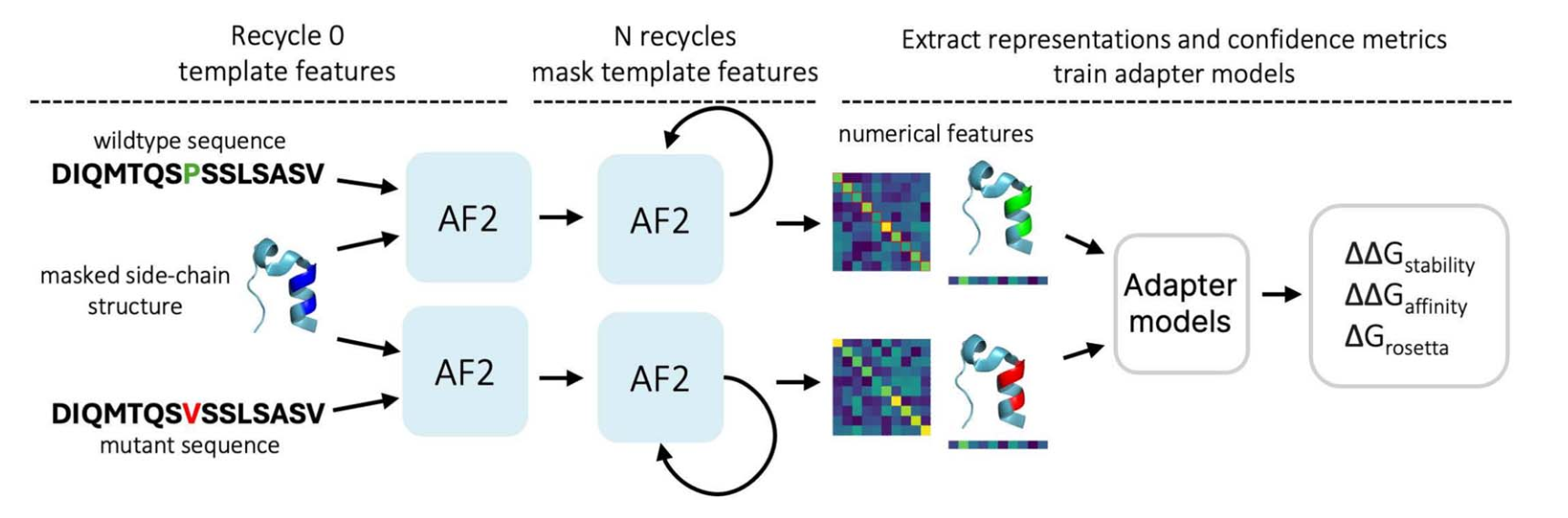
AFToolkit scheme. At each stage of recycling, the framework applies a mask to the structural information of the original protein. For mutation-related tasks, side chain masking is applied.
The article describing the framework was published in Briefings in Bioinformatics. The authors have also made the code for running, training, and their pretrained models available in their repository.